|
Strong-field electronic dynamics is a key ingredient in attosecond science.
Most of theoretical studies of strong-field processes, however, are limited to
the single active electron model, usually considering the highest-occupied
molecular orbital (HOMO) only. For accurate strong-field electronic dynamics
calculations, self-interaction-free time-dependent density-functional theory (TDDFT) is employed taking into account the
detailed electronic structure and multielectron responses [J. Chem. Phys. 123, 062207 (2005)]. By means of the new developed
time-dependent Voronoi-cell finite difference (TDVFD) method on multicenter molecular grids, detailed TDDFT
studies are performed for multiphoton ionization (MPI), photoelectron angular
distribution (PAD), and high-order harmonic generation (HHG) of several
polyatomic molecules in intense linearly-polarized and ultrashort lasers fields
with arbitrary field–molecule orientation. The TDDFT results demonstrate
the importance of multielectron effects on the strong-field multiphoton
processes, such as electron correlation and multiple orbital contributions.
For CO2, the TDDFT results [1] show the maximum peak at 40 degree
for the first time in good agreement with the recent experimental data
regarding the orientation dependence of MPI, whereas significant discrepancy
exists between the experimental data and MO-ADK that is one of widely used
single active electron models. Also PAD of CO2 reveals the delicate
relation between the orientation dependence and the molecular orbital symmetry
[1]. For H2O, the TDDFT results provide the first prediction that
the contribution from the inner orbital below HOMO (HOMO-1) becomes dominant in
the overall patten of the orientation dependence of MPI [2]. This feature
suggests a new way to selectively probe individual molecular orbitals in
strong-field processes. The presented method can be a promising tool to
investigate multielectron effects on attosecond electronic dynamics in
polyatomic molecules.
|
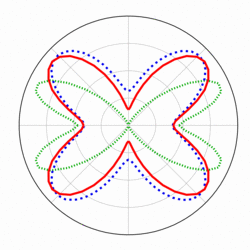
Orientation dependence of CO2 MPI.
Red: TDDFT [1],
Blue: experimental data [J. Phys. Chem. A 112, 9382 (2008)],
Green: MO-ADK [J. Mod. Opt. 54, 967 (2007)].
|
![[bib]](/biblio/img/bibtex.gif) [BibTeX]
[BibTeX]![[pdf]](/biblio/img/pdf.gif) [pdf]
[pdf]![[abstract]](/biblio/img/abstract.gif) [abstract]
[abstract]![[link]](/biblio/img/link.gif) doi:10.1103/PhysRevA.80.011403
doi:10.1103/PhysRevA.80.011403![[slide]](/biblio/img/slide.gif) [slide: 3Mb]
[slide: 3Mb]